
Single-Molecule Scanning Tunnelling Spectroscopy (STS)
The electronic structure of an isolated Single-Ion Molecule Magnet (SIMM) bis(phthalocyaninato) terbium(III) molecule supported on a Cu(111) surface has been investigated by density functional theory (DFT) and scanning tunnelling spectroscopy (STS). These studies reveal that the interaction with the metal surface preserves both the molecular structure and the large spin magnetic moment of the metal center. The 4f electron states are not perturbed by the adsorption while a strong molecular/metal interaction can induce the suppression of the minor spin contribution delocalized over the molecular ligands. The calculations show that the inherent spin magnetic moment of the molecule is only weakly affected by the interaction with the surface.
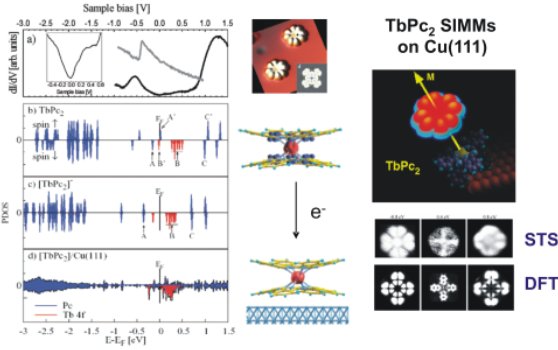
Density of states of TbPc2 molecule on Cu(111). (a) Experimental dI/dV spectrum for the surface-supported molecule measured at the dot position in Figure right. The spectrum achieved on the clean Cu substrate (gray line). In inset, the dI/dV spectrum achieved on the molecule has been replotted to better visualize the low energy range. (b) Calculated spin-resolved electron DOS for the unsupported neutral TbPc2 and (c) charged [TbPc2]- molecule (d) as well as molecular projected DOS (PDOS) for the artificial model Cu/TbPc2 system.
